Le Gène ACO2
Le gène ACO2 et ses mutations

Certaines mutations du gène ACO2 peuvent entraîner des maladies génétiques rares et invalidantes
Le gène ACO2, qui se situe sur le chromosome 22 (locus q13.2), permet la fabrication d'une protéine, appelée aconitase, présente dans les mitochondries, et qui joue un rôle clé dans la production de l'énergie indispensable au bon fonctionnement des cellules.
Lorsque ce gène est défectueux - ou muté - cela peut entraîner des maladies mitochondriales rares et graves qui ralentissent le métabolisme et impactent le bon fonctionnement des organes vitaux ayant le plus besoin d'énergie: les yeux, le cerveau, les muscles, le cœur, le foie, les reins,... Il n'existe aujourd'hui aucun traitement à ces maladies.
Des variants pathogènes du gène ACO2 peuvent causer deux principales formes de maladies rares:
-
les neuropathies optiques héréditaires (NOH), maladies cécitantes, caractérisées par une dégénérescence des cellules ganglionnaires de la rétine, et d'origine mitochondriale: l'atrophie optique dominante (AOD) et l'atrophie optique récessive (AOR), plus rare et plus sévère. Le deuxième gène le plus fréquemment retrouvé dans les AOD est le gène ACO2, après OPA1.
-
la dégénérescence cérébello-rétinienne infantile (ICRD), présentation clinique syndromique grave, associant une atteinte visuelle sévère avec d’autres troubles neurologiques handicapants.
134 mutations uniques du gène ACO2 ont été identifiés et sont recensés dans une base de données globale.


Les maladies rares
Une maladie est dite rare lorsqu'elle affecte moins d'une personne sur 2000. Les maladies rares concernent 3 millions de personnes en France, soit 1 Français sur 20, majoritairement des enfants. Plus de 300 millions de personnes en sont atteintes dans le monde, 30 millions en Europe. Il existe plus de 8000 maladies rares recensées.
Les trois-quarts sont d’origine génétique, et 7 sur 10 débutent dès l’enfance. La grande majorité de ces maladies sont si rares qu’elles sont totalement ignorées du public, voire des médecins. 50 % de ces maladies ne sont pas diagnostiquées, et lorsqu’elles le sont, c’est le plus souvent après des années d’errance diagnostique. Elles sont souvent chroniques, mettant en jeu le pronostic vital, et sont dites "orphelines" car il existe peu ou pas de traitement du tout. C'est pour cela que les maladies génétiques rares constituent un véritable défi de santé publique, et ont un un besoin si crucial et urgent de financements.
Leur étude apporte également des connaissances générales en biologie et en médecine, très précieuses car elles contribuent à mieux comprendre des mécanismes applicables ou transposables à des maladies communes, comme certains cancers ou même les formes graves de Covid-19.
La recherche sur les maladies rares aura par exemple fortement contribué à la découverte de vaccins contre le COVID-19, en particulier ceux basés sur la technologie de l'ARN messager.
Le gène ACO2 et ses fonctions
Le gène ACO2 code la protéine mitochondriale Aconitase qui contribue au cycle de Krebs en isomérisant le citrate en iso-citrate, par l'intermédiaire du cis-aconitate.
Les mutations du gène ACO2 peuvent êtres héritées d'un ou deux parents porteurs sains, ou survenir "de novo" en raison d'une erreur dans la réplication cellulaire.
Certaines mutations du gène ACO2 peuvent générer un déficit dans l'activité enzymatique de l'aconitase, essentielle au bon fonctionnement des mitochondries, du Cycle de Krebs, et donc à la production d'énergie des cellules.
ACO2 est également appelé aconitase 2, ACONM, ICRD, OCA8, HEL-S-284, OPA9.
Lire la description détaillée de la fonction du gène ACO2 sur OMIM (#100850);
Un gène (ACO2), deux phénotypes (OPA9, ICRD):
Mots clés: ACO2, aconitase 2, mitochondria, krebs cycle, optic neuropathy, infantile cerebellar-retinal degeneration, ICRD, cerebellar atrophy, mitochondrial dysfunction, neurometabolic disorders

.png)
Vivre avec une mutation du gène ACO2
Les maladies causées par des mutations du gène ACO2 peuvent s'exprimer par un ou plusieurs des symptômes suivants, avec des niveaux de sévérité et d'évolutivité très variables d'un patient à l'autre:
-
Atrophie optique isolée, pour les formes les moins graves
-
Acuité visuelle réduite voire nulle
-
Vision périphérique principalement (scotome paracentral)
-
Nystagmus, strabisme
-
Rétinopathie pigmentaire
-
Forte sensibilité à la lumière
-
Hypotonie, scoliose
-
Grande fatigabilité
-
Important retard des acquisitions et du développement
-
Déficience intellectuelle
-
Ataxie
-
Microcéphalie
-
Anomalies de la substance blanche du cerveau à l'IRM
-
Possible épilepsie
-
Troubles de la digestion, difficultés à s'alimenter
Dans le cas des formes syndromiques les plus sévères affectant le cerveau (ICRD - Infantile Cerebellar Retinal Degeneration), les premiers symptômes apparaissent dès les premiers mois de l'enfance. Elles peuvent être graves, handicapantes, et potentiellement évolutives.
Pour établir un diagnostic, il est important de consulter neuropédiatre et ophtalmologue. Des recherches génétiques peuvent s'avérer nécessaires après l'évaluation clinique, pour confirmer le gène en cause.
Aucun traitement n'existe à ce jour. Les prises en charge reposent sur un suivi multi-disciplinaire: orthoptie et rééducation basse vision pour favoriser le développement d’une vision excentrée, orthophonie, kinésithérapie, psychomotricité, ergothérapie, et un suivi médical régulier et personnalisé.
Les protocoles nationaux de diagnostic et de soins (PNDS) liés aux NOH (Neuropathies optiques héréditaires) viennent de sortir et ont été mis en ligne sur le site de la Haute autorité de Santé. Ces documents jouent un rôle majeur dans la prise en charge des patients. Ils ont pour objectif d’expliciter aux professionnels concernés (médecins généralistes, médecins spécialistes, médecins de la Sécurité Sociale etc…) et aux patients la prise en charge diagnostique et thérapeutique optimale actuelle et le parcours de soins d’un patient atteint d’une maladie rare donnée en s’appuyant sur les recommandations internationales déjà publiées. Ils sont rédigés par les experts des Centres de Référence Maladies Rares (CRMR) à l’aide d’une méthodologie proposée par la Haute Autorité de Santé (HAS).
Les PNDS relatifs aux NOH ont été publiés en juillet 2021 par le Centre de Référence des maladies rares en ophtalmologie OPHTARA et relus par les associations de patients concernées, dont ACO2 GENE.
Le gène ACO2 est alors appelé OPA 9 lorsque la pathologie est une neuropathie optique. Accéder aux revues de la littérature (focus sur OPA9 en page 22).
L'ADN, nos chromosomes, nos gènes
Notre corps est constitué de milliards de cellules. Chaque cellule possède un noyau constitué de chromosomes contenant de l’ADN, notre code génétique, qui indique au corps comment se développer et fonctionner.
Les chromosomes contiennent des informations génétiques qui se regroupent en gènes. Chaque gène, lorsqu'il fonctionne correctement, a un rôle spécifique dans le corps.
Des mutations génétiques provoquant une mauvaise synthèse des protéines peuvent survenir sur cette séquence d'ADN. Le gène, endommagé, peut donner lieu à une cellule défaillante et ainsi être le point de départ d’une maladie génétique ou d'un cancer.

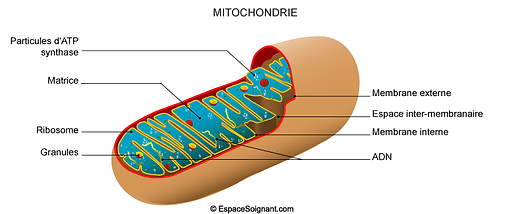
Les mitochondries
Les mitochondries constituent un élément essentiel dans la production d’énergie nécessaire au fonctionnement de nos cellules. Elles sont impliquées dans plusieurs fonctions essentielles, dont la principale est la production d'ATP (énergie) et sont souvent appelées "centrales énergétiques de nos cellules".
Les mitochondries sont des organites situés à l’intérieur de la cellule, d’une taille avoisinant un micromètre de longueur. Elles contiennent des enzymes qui accélèrent les réactions de transformation des aliments en énergie, liées à une chaîne complexe : la chaîne respiratoire.
Les mitochondries produisent ainsi 90% de l’énergie cellulaire dont les tissus, les organes et l’organisme entier ont besoin pour fonctionner. Un seul déficit de ces enzymes, tels que l'aconitase par exemple, peut entraîner un dysfonctionnement des mitochondries ainsi que des symptômes très graves et variés qui vont agir sur nos organes vitaux.
Les maladies mitochondriales constituent un large groupe de maladies qui peuvent affecter des sujets de tout âge avec une prévalence d’environ 1 sur 4000 naissances soit près de 200 nouveaux cas chaque année en France. Les pathologies associées sont très complexes et diverses, affectent plusieurs organes, présentant parfois un seul symptôme, mais en combinant souvent de multiples. Plus de 300 gènes sont impliqués dans les maladies mitochondriales.
"C'est quoi, la mitochondrie ?"
par Vincent Procaccio
Professeur de génétique à l'Université d'Angers, Vincent Procaccio travaille sur le diagnostic des maladies mitochondriales, la connaissance de ces maladies complexes qui touchent principalement les organes les plus fortement consommateurs d’énergie – le muscle, le cerveau, l’œil et l’identification de nouveaux traitements.
Liens utiles et publications
Liens vers les principales publications scientifiques sur le gène ACO2:
-
Infantile Cerebellar-Retinal Degeneration associated with a mutation in mitochondrial aconitase (R.Spiegel, 2012)
-
Mutations in the tricarboxylic acid cycle enzyme, aconitase 2, cause either isolated or syndromic optic neuropathy with encephalopathy and cerebellar atrophy (M.Metodiev, 2014)
-
Clinical, radiological, and genetic characteristics of 16 patients with ACO2 gene defects: Delineation of an emerging neurometabolic syndrome (R.Sharkia, 2018)
-
ACO2 mutations: A novel phenotype associating severe optic atrophy and spastic paraplegia (C.Marelli, 2018)
-
Identification of novel compound heterozygous mutations in ACO2 in a patient with progressive cerebral and cerebellar atrophy (M.Fukada, 2019)
-
Derivation of a human DOA iPSC line, IISHDOi006-A, with a mutation in the ACO2 gene (V.Cerrada, 2019)
-
Novel compound heterozygous ACO2 mutations in an infant with progressive encephalopathy: A newly identified neurometabolic syndrome (JS.Park, 2020).
-
Recessive ACO2 variants as a cause of isolated ophthalmologic phenotypes (S.Gibson, 2020).
-
Expanding the clinical and phenotypic heterogeneity associated with biallelic variants in ACO2 (PR Blackburn, 2020).
-
Haploinsufficiency due to a novel ACO2 deletion causes mitochondrial dysfunction in fibroblasts from a patient with dominant optic nerve atrophy (MAC Neumann, 2020)
-
Complex hereditary spastic paraplegia associated with episodic visual loss caused by ACO2 variants (T.Tozawa, 2021)